According to different risk levels, FDA classifies medical devices into three categories (ⅰ, ⅱ, ⅲ), of which class I has the lowest risk level and class iii has the highest risk level.
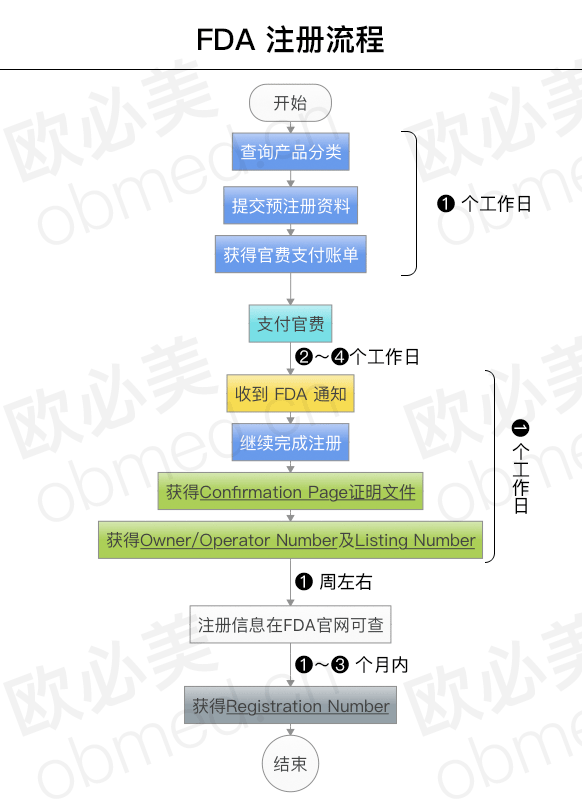
For class I products (about 47%), the implementation of General Control, to comply with GMP standards, most of the products only need to be registered and listed, products can enter the United States market.
For class ⅱ products (accounting for about 46%), the implementation of Special Control, to comply with GMP standards, enterprises after registration and listing, but also need to submit 510(K) application (510(K) exemption for very few products, please contact the FDA experts to understand the details of the category. Tel: 400-686-5670), it is the sales recognition of the second class products.
For class iii products (accounting for about 7%), pre-market approval is implemented. After registration and listing, enterprises are required to implement GMP standards and submit PMA (Premarket Application) applications to FDA (part of class III products are PMN applications).
For class ⅰ and ⅱ devices, some products are exempted from GMP audit, while for class ⅲ devices, enterprises must submit PMN or PMA. The FDA's official Clearance, which accompanies the announcement, allows companies to market their products under their own name in the U.S. medical device market. Whether to conduct on-site GMP assessment during the application process is determined by FDA based on a combination of factors such as product risk level, management requirements and market feedback.